Para la creación de una "Metodología de Trabajo" que enseñe acerca de los priones, se debe hacer mediante la creación de una síntesis bien elaborada de este tema, que resuma todo y además tenga el apoyo de ilustraciones que favorezcan la posibilidad de entender este tema.
En algunos casos sería bueno también, que alguno de los diferentes subtópicos de este tema tenga ademas, el apoyo de tablas comparativas cotejando diferentes cosas y de flujogramas o mapas conceptuales, que sean un tipo de ayuda didáctica en el ordenamiento de las distintas fases que comprende dicho tema.
En caso de tener que realizarse, algún tipo de exposición o presentación, la mejor manera sería mediante el uso de herramientas como lo es un documento de power point (.ppt).
lunes, 9 de mayo de 2011
lunes, 2 de mayo de 2011
Novena Entrada: Fuente de Información como Apoyo a Priones.
VIDEOS DE MUCHO INTERÉS
2) YOUUTUBE. Human prion protein in 3-D showing high degree of alpha-helical structure [En Línea]. <http://www.youtube.com/watch?v=w8-MAp4B2D4&feature=related> [Citado el 2 de Mayo de 2011].
3) YOUTUBE. Prion propagation [En Línea]. <http://www.youtube.com/watch?v=pMijlD8YEtQ&feature=related> [Citado el 2 de Mayo de 2011].
4) YOUTUBE. Prions [En Línea]. <http://www.youtube.com/watch?v=m4swohcGWAM&feature=related> [Citado el 2 de Mayo de 2011].
lunes, 25 de abril de 2011
Octava Entrada: Evaluación de Sitios WEB sobre Priones.
URL #1: http://www.veterinaria.org/revistas/vetenfinf/bse/priones/priones.htm
Validez:
Buena información y gran profundización en el tema.
Pertinencia:
Resulta cómodo y coherente.
Confiabilidad:
Fue publicado por una importante revista, tiene una buena bibliografía y una correcta citación.
Relevancia:
Es un excelente medio de consulta.
Actualidad o Vigencia y Referencia de las mismas:
Tiene información muy contemporánea.
URL #2: http://usuarios.multimania.es/priones/
Validez:
Mucha investigación y buen desarrollo de los conceptos.
Pertinencia:
Es muy especifico y tiene una gran síntesis del tema.
Confiabilidad:
Hecho por un estudiante de biología, tiene una buena bibliografía y una correcta citación.
Relevancia:
Es una gran ayuda para aprender.
Actualidad o Vigencia y Referencia de las mismas:
Tiene información muy contemporánea.
Validez:
Buena información y gran profundización en el tema.
Pertinencia:
Resulta cómodo y coherente.
Confiabilidad:
Fue publicado por una importante revista, tiene una buena bibliografía y una correcta citación.
Relevancia:
Es un excelente medio de consulta.
Actualidad o Vigencia y Referencia de las mismas:
Tiene información muy contemporánea.
URL #2: http://usuarios.multimania.es/priones/
Validez:
Mucha investigación y buen desarrollo de los conceptos.
Pertinencia:
Es muy especifico y tiene una gran síntesis del tema.
Confiabilidad:
Hecho por un estudiante de biología, tiene una buena bibliografía y una correcta citación.
Relevancia:
Es una gran ayuda para aprender.
Actualidad o Vigencia y Referencia de las mismas:
Tiene información muy contemporánea.
lunes, 18 de abril de 2011
Séptima Entrada: Articulo de Revista sobre Priones.
Titulo: Enfermedades por priones: de la clínica a la biología molecular
URL: http://www.acnweb.org/acta/acta_2010_26_2_87-111.pdf
Referencia bibliográfica: COLLINGE J. Prion diseases of humans and animals: their causes and molecular basis. Annu Rev Neurosci 2001; 24: 519-550.
La importancia de esta referencia bibliográfica, se da porque es un importante articulo de revista indexada, el cual es de gran ayuda para la redacción de este importante articulo de revista. En este se habla de las enfermedades priónicas, de su causa y su base molecular.
URL: http://www.acnweb.org/acta/acta_2010_26_2_87-111.pdf
Referencia bibliográfica: COLLINGE J. Prion diseases of humans and animals: their causes and molecular basis. Annu Rev Neurosci 2001; 24: 519-550.
La importancia de esta referencia bibliográfica, se da porque es un importante articulo de revista indexada, el cual es de gran ayuda para la redacción de este importante articulo de revista. En este se habla de las enfermedades priónicas, de su causa y su base molecular.
lunes, 11 de abril de 2011
lunes, 4 de abril de 2011

lunes, 28 de marzo de 2011
Cuarta Entrada: Enfermedad del Alzheimer (EA), tema asociado a Priones.
Prion, ¿partícula proteica estable o transformación dinámica? Un nuevo enfoque sobre las enfermedades priónicas y la enfermedad de Alzheimer
RESUMEN
En este trabajo se comparan las enfermedades priónicas con la enfermedad de Alzheimer, tratando de dar una explicación, en el orden epidemiológico, al aumento de la incidencia de esta última, así como la similitud que se establece en el ámbito clínico-patológico, la cual no obedece a la casualidad y sí probablemente a la causalidad en cuanto a mecanismos comunes en su origen, pues son los trastornos en la conformación de la estructura proteica normal lo que pudiera explicar no sólo la ocurrencia de estas enfermedades, sino también la capacidad de tornarse infecciosas, con la potencialidad de ser contagiosas en mayor o menor período y en mayor o menor grado.
Palabras clave: Prión, enfermedades priónicas, enfermedad de Alzheimer, transformación proteica infecciosa, escala evolutiva.
INTRODUCCIÓN
Las proteínas constituyen un componente esencial de las células y tienen una función bien definida en relación con la estructura conferida por mecanismos genéticamente determinados; las alteraciones estructurales producidas en los procesos de plegamiento de algunas proteínas intracelulares que dan origen a modificaciones funcionales, son quizás la base de un nuevo enfoque en la visión de las llamadas enfermedades priónicas y de otras como la enfermedad de Alzheimer (EA) con la cual existen similitudes que no deben ignorarse.1
Se han señalado semejanzas en lo que respecta a la acumulación de proteína amiloide en un grupo de enfermedades entre las cuales se encuentran la EA y las enfermedades priónicas agrupadas bajo la clasificación de enfermedades conformacionales, aunque las alteraciones proteicas estructurales en el orden del plegamiento son diferentes como disímiles sus expresiones clínico-patológicas.2
Hasta el presente, sólo algunas enfermedades por priones tienen probada su infectividad siguiendo patrones clínicos particulares; desde que Prusiner identificó y definió el término prión,3 numerosos investigadores han señalado sólo un número limitado de esta “élite” entre las cuales se encuentran la enfermedad de Creutzfeldt-Jakob (ECJ), algunas que afectan al ser humano y otras formas de encefalopatías del ámbito veterinario, algunas de las cuales son transmisibles a la especie humana.4,5 Las enfermedades conformacionales2,6 pudieran ser un eslabón entre las alteraciones estructurales y la adquisición de cierto grado de infectividad en las enfermedades priónicas y la EA.
DESARROLLO
Los cambios en la estructura proteica incluyen un proceso de nucleación, en el cual existen oligomeros activos que funcionan como una "semilla" para convertir una conformación normal en la isoforma anormal, estas proteínas con trastornos en el plegamiento son producidas con frecuencia en el citoplasma o el retículo endoplásmico, este proceso está ocurriendo de forma continua en la célula, donde sucede una degradación proteosomal, existen proteínas que actúan previniendo la acumulación de aquellas mal plegadas y el desarrollo de enfermedades por esta causa.
Al fallar estos mecanismos, ocurre acumulación de proteínas no plegadas prefibrilares que forman agregados intracelulares y resultan citotóxicos.7,8 Tal vez, la presencia en cuanto a calidad o cantidad de estos agregados prefibrilares intervienen de forma directa en la capacidad para tornar una proteína normal en infecciosa y potencialmente contagiosa, esto se ha comprobado en cierto grado en la amiloidosis sistémica,9 lo que parece indicar la relación entre los cambios conformacionales y el carácter infeccioso de algunas proteínas; trabajos recientes sugieren que las enfermedades por priones pueden ser consideradas como una forma transmisible de amiloidosis cerebral.2,9,10 Otros autores sin embargo, no lo consideran así porque no resulta claro si los agregados presentes en estas entidades están implicados en la neurodegeneración de forma directa o si sólo son una consecuencia de la misma.2
El término prión tendría sentido como adquisición del carácter infeccioso de una proteína celular y pudiera ser empleado como sinónimo de transformación o cambio proteico infeccioso.
Aunque la proteína amiloide es diferente en muchos casos en cuanto a aspecto, tamaño, secuenciación o estructura nativa, todas ellas forman agregados fibrilares que muestran un marcado parecido en cuanto a morfología, estructura interna y a la presencia de aquellas proteínas llamadas serpinas, las cuales son un elemento común entre virus, células eucariotas y procariotas.11 Cambios en el pH, temperatura o exposición a ciertos agentes químicos pueden producir rupturas y cambios en la proteína original para formar proteína amiloide y, en muchos casos, estructuras fibrilares.12
El depósito de proteínas anormales con una u otra conformación produce enfermedades con deterioro cognitivo, trastornos conductuales, convulsiones y tienen un curso fatal; en el orden anatomopatológico se ha observado la acumulación de proteínas anormales en cuanto a su conformación y función, formación de placas amiloides y ovillos neurofibrilares y cambios degenerativos al nivel del núcleo de Meynert.13-15
Se desconoce a ciencia cierta hasta qué punto la EA y las llamadas enfermedades priónicas representan un peligro en el orden epidemiológico-infeccioso, es posible que constituyan en el futuro epidemias devastadoras por sus mecanismos de transmisión y su carácter solapado.
En el caso de las enfermedades priónicas como la ECJ, se invocan las mutaciones peptídicas de tal forma que se produce una estructura beta plana que ha sido demostrada como infecciosa, la llamada proteína priónica.1,15,16
Cualesquiera que sean los mecanismos mediante los cuales estas proteínas producidas se comportan como infecciosas, la no demostración del carácter definitivamente contagioso de la EA hasta este momento, pudiera deberse al largo período de latencia que ocurre desde la adquisición de la proteína infecciosa hasta el desarrollo de los primeros síntomas o signos de la enfermedad en el orden clínico, los cuales, por lo general, demoran años en manifestarse, si en un inicio las enfermedades producidas por priones fueron llamadas enfermedades por virus lentos17 fue precisamente por lo tardío de la expresión clínica con respecto al momento de la adquisición del contagio, ahora, con respecto a la EA, ¿acaso no pudiera tratarse de otra enfermedad producida por "virus más lentos"? usando los antiguos y ya caducos términos para una mejor comprensión del problema. Por otra parte, la coexistencia de demencia de causa vascular con la EA pudiera enmascarar los síntomas iniciales de esta última; además muchos ancianos mueren sin que se les realice un estudio mas allá del rutinario para establecer tal diferencia.
La incidencia de la EA en el mundo entero pudiera ser mucho más grande que lo que ha sido publicado, pues hay numerosos países que no contemplan este tema entre los objetivos priorizados de su sistema de salud.18-20
El surgimiento del concepto de proteína amiloide infecciosa dentro del contexto de las enfermedades conformacionales con expresiones clínicas diversas pudiera ser de importancia, pues ello pudiera incluir lo que de forma más estrecha se llama proteína priónica en las enfermedades por priones conocidas o proteina tau en la EA.
Quizá los cambios proteicos infecciosos sean diferentes tipos de respuestas de las proteínas del sistema nervioso a una gama limitada de agentes causales, determinando su expresión en el orden clínico-patológico y según las particularidades del código genético que les da origen, manifestándose unas veces como cualquiera de las enfermedades priónicas conocidas y otras veces de manera diferente como la EA, aunque esta hipótesis parece ser menos probable.
En un ordenamiento evolutivo decreciente, de los hongos a las bacterias, de estas a los virus y de estos últimos a los cambios o transformaciones proteicas infecciosas que pudieran constituir el elemento más primitivo de esta secuencia, parecen existir leyes que rigen el comportamiento y capacidad de cada escalón para producir enfermedades, pues mientras más primitiva es su estructura, mayor es el daño biológico que causan, más confusos resultan sus mecanismos productores de daño, más difíciles son de reconocer epidemiológicamente debido al poder de mutabilidad de unos o el mimetismo de otros, los cuales llegan al extremo de parecerse a proteínas celulares normales y evadir el sistema inmune quien no los reconoce en muchos casos como extraños aún habiendo variado su conformación.
¿Pero acaso proteínas aparentemente inocuas, tienen la capacidad de transformarse en infecciosas bajo determinadas condiciones o son un grupo de ellas las que lo hacen? La respuesta a esta interrogante se desconoce aún, pero pudiera ser un buen punto de partida para investigaciones posteriores.
En conclusión, la adquisición del carácter infeccioso de una proteína celular pudiera deberse a cambios en su conformación en relación con aspectos ambientales y genéticos, tanto para la EA como en las enfermedades priónicas, la carencia de pruebas del carácter infecto-contagioso de la EA tal vez se deba a su largo período de latencia y a lo confuso de sus diferencias con otras formas de demencia, es posible que las transformaciones proteicas infecciosas sean un eslabón evolutivo dinámico, pero inferior en relación con los virus, hongos y bacterias, de ahí la variabilidad en su expresión clínica.
REFERENCIAS BIBLIOGRÁFICAS
1. Velayos JL, Irujo A, Cuadrado-Tejedor M, Paternain B, Moleres FJ, Ferrer V. The cellular prion protein and its role in Alzheimer's disease. Prion. 2009;3:110-7.
2. Lupi O, Peryassu M. An emerging concept of prion infections as a form of transmissible cerebral amyloidosis. Prion. 2007;1:223-7.
3. Prusiner SB, Hsiao KK. Human prion diseases. Ann Neurol. 1994;35:385-95.
4. Zivkovic S, Boada M, López O. Revisión de la enfermedad del Creutzfeldt-Jakob y otras enfermedades priónicas. Rev Neurol. 2000;31:1171-9.
5. Biacabe AG, Jacobs JG, Bencsik A, Langeveld J, Baron T. H-type bovine spongiform encephalopathy. Complex molecular features and similarities with human prion diseases. Prion. 2007;1:61-8.
6. Mena M, Rodríguez-Navarro J, García de Yébenes J. The multiple mechanisms of amyloid deposition. The role of parkin. Prion. 2009;3:5-11.
7. Stefani M, Dobson CM. Protein aggregation and aggregate toxicity: new insights into protein folding, misfolding diseases and biological evolution. J Mol Med. 2003;81:678-99.
8. Gregersen N, Bross P, Andresen BS, Pedersen CB, Corydon TJ, Bolund L. The role of chaperone-assisted folding and quality control in inborn errors of metabolism: Protein folding disorders. J Inherit Metab Dis. 2001;24:189-212.
9. Lundmark K, Westermark GT, Nystrom S, Murphy CL, Solomon A, Westermark P. Transmissibility of systemic amyloidosis by a prion-like mechanism. Proc Natl Acad Sci USA. 2002;99:6979-84.
10. Maddelein ML. Infectious fold and amyloid propagation in podospora anserina. Prion. 2007;1:44-7.
11. Belorgey D, Hägglöf P, Karlsson-Li S, Lomas D. Protein Misfolding and the Serpinopathies. Prion. 2007;1:15-20.
12. Dickson DW, Crystal HA, Bevona C, Honer W, Vincent I, Davies P. Correlations of synaptic and pathological markers with cognition of the elderly. Neurobiol Aging. 1995;16:285-98.
13. Jahn TR, Radford SE. The Yin and Yang of protein folding. FEBS J. 2005;272:5962-70.
14. Guimerà A, Gironès X, Cruz-Sánchez F. Actualización sobre la patología de la enfermedad de Alzheimer. Rev Esp Patol. 2002;35:21-48.
15. Ariza A. El patólogo ante las encefalopatías espongiformes transmisibles. Rev Esp Patol. 2002;35:49-62.
16. Hamilton JA, Whitty G, White AR, Jobling MF, Thompson A, Barrow CJ. Alzheimer's disease amyloid beta and prion protein amyloidogenic peptides omote macrophage survival, DNA synthesis and enhanced proliferative response to CSF-1 (M-CSF). Brain Res. 2002;940:49-54.
17. Fernández MI, Arribas MA, Fernández Buey N. Demencias por virus lentos: enfermedades tipo prión (encefalopatías espongiformes transmisibles). Medicine. 1998;7:4470-3.
18. Alzheimer's Disease Education and Referral Center. Progress report on Alzheimer's disease: taking the next steps. Silver Spring, MD: 2001. (NIH Publication No. 00-4859).
19. Petersen RC. Practice parameter: early detection of dementia: mild cognitive impairment (an evidence-based review), Report of the Quality Standards Subcommittee of the American Academy of Neurology. Neurology. 2001;56:1133-42.
20. National Center for Health Statistics. Deaths and death rates for the 10 leading causes of death in specified age groups, by race and sex: United States, 1998. National Vital Statistics Reports. 2000;48(11):27-9.
Dr. Edmundo Rivero Arias. Hospital Clinicoquirúrgico "Hermanos Ameijeiras", San Lázaro No. 701 entre Belascoaín y Marqués González, Centro Habana, Ciudad de La Habana, Cuba. CP 10300. Correo electrónico:erivero@infomed.sld.cu
BIBLIOGRAFÍA
RIVERO ARIAS, Edmundo. Prion, ¿partícula proteica estable o transformación dinámica? Un nuevo enfoque sobre las enfermedades priónicas y la enfermedad de Alzheimer. Rev cubana med [online]. 2010, vol.49, n.3, pp. 262-267. ISSN 0034-7523.
lunes, 21 de marzo de 2011
Tercera Entrada: Sinónimos, Acrónimos o Variantes de la palabra Prión.
PRION: PROTEINACEOUS INFECTIOUS AGENT, PROTEINAS PRPC Y PROTEINAS PRPSC.
URL PALABRA PRION:
1. es.wikipedia.org/wiki/Prion
2. ciencia.glosario.net/biotecnologia/prión-10166.html
3. svneurologia.org/congreso/priones-1.html
4. www.enciclonet.com/articulo/prion/
5. http://bvs.sld.cu/revistas/san/vol13_1_09/san08109.htm
URL SINÓNIMOS, ACRÓNIMOS O VARIANTES PALABRA PRION:
1. medind.nic.in/jac/t03/i4/jact03i4p334.pdf
2. encyclopedia.farlex.com/Prion+(infectious+agent)
3. http://etd.ohiolink.edu/send-pdf.cgi/CYRANEK%20WIOLETA.pdf?csu1236370744
4. www.ncbi.nlm.nih.gov/pubmed/15240887
5. http://serendip.brynmawr.edu/biology/b103/f97/projects97/Butler.html
URL PALABRA PRION:
1. es.wikipedia.org/wiki/Prion
2. ciencia.glosario.net/biotecnologia/prión-10166.html
3. svneurologia.org/congreso/priones-1.html
4. www.enciclonet.com/articulo/prion/
5. http://bvs.sld.cu/revistas/san/vol13_1_09/san08109.htm
URL SINÓNIMOS, ACRÓNIMOS O VARIANTES PALABRA PRION:
1. medind.nic.in/jac/t03/i4/jact03i4p334.pdf
2. encyclopedia.farlex.com/Prion+(infectious+agent)
3. http://etd.ohiolink.edu/send-pdf.cgi/CYRANEK%20WIOLETA.pdf?csu1236370744
4. www.ncbi.nlm.nih.gov/pubmed/15240887
5. http://serendip.brynmawr.edu/biology/b103/f97/projects97/Butler.html
lunes, 14 de marzo de 2011
Segunda Entrada: Enfermedades por Priones, el PrPC y el PrPSc
FUENTE ORAL
FUENTE ESCRITA
¿Qué esperaba encontrar en esta fuente?
Encontrar las diferentes enfermedades causadas por priones, las que se dan en humanos y las que se dan en animales. saber como se dan, cuál es el mecanismo patológico, qué tan frecuentes son estas enfermedades, qué es lo que sucede a nivel molecular y qué se puede hacer clínicamente.
¿Qué encontró?
Las enfermedades ocasionadas por priones son también conocidas como encefalitis espongiformes transmisibles o demencias de tipo infeccioso. En humanos las presentaciones clínicas más reconocidas son la enfermedad de Creutzfeldt-Jakob, el síndrome de Gerstmann-Sträussler-Scheinker y el insomnio fatal familiar. Son consideradas patologías poco comunes, pero los descubrimientos en biología molecular de los últimos años muestran que los mecanismos patológicos que llevan a su desarrollo pueden ser comunes a varias enfermedades neurodegenerativas, lo cual puede ampliar el espectro patológico, convirtiéndolas en alteraciones no tan infrecuentes en neurología.
Esta revisión pretende dar herramientas al clínico para reconocer estas enfermedades discutiendo las presentaciones clínicas en seres humanos con sus variantes: esporádicas, infecciosas y familiares, comentando además el uso de laboratorios, criterios diagnósticos y aproximación terapéutica.
¿Qué utilidades tiene esta información para ampliar sus reconocimientos con respecto al tema?
Resultó demasiado constructivo, puesto que esta charla, abrió las puertas a grandes incógnitas. La información suministrada es de gran ayuda para la investigación, logra ubicarse con mas profundidad en el tema y resaltar temas que no se habían tomado con antelación.
De la charla con: Carlos Andrés Villegas Lanau. Médico. Magíster en Ciencias Básicas Biomédicas. Doctorado en Ciencias, Universidad de Antioquia. Profesor del Instituto de Investigaciones Médicas, Facultad de Medicina de la Universidad de Antioquia. Investigador del Grupo de Neurociencias de Antioquia.
Correo electrónico: andres.villegas@neurociencias.udea.edu.co
¿Qué esperaba encontrar en esta fuente?
Encontrar las diferentes enfermedades causadas por priones, las que se dan en humanos y las que se dan en animales. saber como se dan, cuál es el mecanismo patológico, qué tan frecuentes son estas enfermedades, qué es lo que sucede a nivel molecular y qué se puede hacer clínicamente.
¿Qué encontró?
Fue Stanley Prusiner quien desarrolló la hipótesis priónica, postulando que una proteína llamada priónica (PrP) puede adoptar dos configuraciones distintas, y una de estas conformaciones puede llevar al cambio de conformación de la otra proteína, lo cual produce un comportamiento de la proteína que provoca el cambio conformacional, similar al de un agente infeccioso. Prusiner también acuñó el término prión (acróstico de proteinaceous infectious agent), descubrió el gen codificador para la proteína priónica que normalmente se encuentra en las células (PrPC) y desarrolló modelos transgénicos que ayudaron a demostrar su hipótesis.
En general las enfermedades causadas por priones poseen características comunes, como son: pérdida progresiva de neuronas, reducida respuesta inflamatoria, vacuolización del neuropilo (provocando la llamada encefalopatía espongiforme), depósitos de proteína priónica con conformación anómala y la capacidad de transmitir la enfermedad (por lo que también son llamadas encefalopatías espongiformes transmisibles). Además se ha identificado a las enfermedades por priones en humanos como demencia infecciosa, debido al cuadro de demencia rápidamente progresiva que las caracteriza. En esencia, se identifica a un prión como una proteína que por un cambio en su conformación (transformación de hélices alfa en pliegues beta) cambia sus propiedades físico-químicas, haciéndose más resistente a agentes que normalmente la degradarían y formando agregados proteicos; adicionalmente, adquiere la capacidad de provocar en proteínas iguales a ella el mismo cambio conformacional, convirtiéndolas en un agente transmisible, esto es, puede pasar de un individuo a otro. En mamíferos sólo se conoce una proteína con esas características, llamada PrP, pero se especula sobre otras que parecen tener las mismas características.
¿Qué utilidades tiene esta información para ampliar sus reconocimientos con respecto al tema?
Fue muy bueno el documento que encontré para este trabajo, en el encontré una amplia información y una gran ayuda para el desarrollo de mi investigación.
Del documento: Carlos Andrés Villegas Lanau. Enfermedades por priones: de la clínica a la biología molecular. Acta Neurol Colomb 2010;26:87-111.
FUENTE ELECTRÓNICA O VIRTUAL
¿Qué esperaba encontrar en esta fuente?
Encontrar las diferentes enfermedades causadas por priones, las que se dan en humanos y las que se dan en animales. saber como se dan, cuál es el mecanismo patológico, qué tan frecuentes son estas enfermedades, qué es lo que sucede a nivel molecular y qué se puede hacer clínicamente.
¿Qué encontró?
INTRODUCCIÓN
Hace más de 200 años fue descrita en Inglaterra una enfermedad que afecta a ovejas y cabras, y que por su síntoma de rascado intenso, el cual les provoca laceraciones y pérdida de la lana, fue llamada scrapie (rascar, en inglés). Esta enfermedad se acompaña de ataxia progresiva y temblor, y es fatal en todos los casos. Cuando se identificó el scrapie como una enfermedad infecciosa se trató de identificar el agente etiológico causal, encontrando que se trataba de uno filtrable, con un tiempo muy prolongado de incubación y capaz de soportar tratamientos con calor, formaldehído y radiación que normalmente destruyen a los agentes infecciosos conocidos, de manera que fue clasificado inicialmente como virus lento o agente no convencional. El hallazgo de que el agente del scrapie carece de ácidos nucleicos llevó a John Stanley Griffith a proponer que la enfermedad es producida por una proteína que se comporta como un agente infeccioso. Carleton Gajdusek fue el primero en identificar una enfermedad en humanos cuyo agente causal tiene las mismas características que las encontradas en el scrapie; fue identificada como kuru, una patología que afecta a la tribu Fore, la cual habita las tierras altas de Papúa, en Nueva Guinea, e identificó la causa de la transmisión de la enfermedad por un rito caníbal desarrollado en esta comunidad. Estos descubrimientos condujeron a identificar otras patologías, diferentes al kuru, en humanos, causadas por agentes con las mismas características del scrapie, como la enfermedad de Creutzfeldt-Jakob, y posteriormente, el síndrome de Gerstmann-Sträussler-Scheinker.
¿Qué utilidades tiene esta información para ampliar sus reconocimientos con respecto al tema?
La ayuda en formato virtual, también fue un buen sistema de información para el trabajo, en este pude encontrar en gran parte la historia de los inicios de priones, como una enfermedad que afecta a humanos y a animales.
Tomado de: Carlos Andrés Villegas Lanau. Enfermedades por priones: de la clínica a la biología molecular. [En Línea]. http://www.acnweb.org/acta/acta_2010_26_2_87-111.pdf 14 de Marzo de 2011.
lunes, 7 de marzo de 2011
Primera Entrada: Prion, ¿partícula proteica estable o transformación dinámica? Un nuevo enfoque sobre las enfermedades priónicas y la enfermedad de Alzheimer
SINTESIS
En este trabajo se comparan las enfermedades priónicas con la enfermedad de Alzheimer, tratando de dar una explicación, en el orden epidemiológico, al aumento de la incidencia de esta última, así como la similitud que se establece en el ámbito clínico-patológico, la cual no obedece a la casualidad y sí probablemente a la causalidad en cuanto a mecanismos comunes en su origen, pues son los trastornos en la conformación de la estructura proteica normal lo que pudiera explicar no sólo la ocurrencia de estas enfermedades, sino también la capacidad de tornarse infecciosas, con la potencialidad de ser contagiosas en mayor o menor período y en mayor o menor grado.
INTRODUCCIÓN
Las proteínas constituyen un componente esencial de las células y tienen una función bien definida en relación con la estructura conferida por mecanismos genéticamente determinados; las alteraciones estructurales producidas en los procesos de plegamiento de algunas proteínas intracelulares que dan origen a modificaciones funcionales, son quizás la base de un nuevo enfoque en la visión de las llamadas enfermedades priónicas y de otras como la enfermedad de Alzheimer (EA) con la cual existen similitudes que no deben ignorarse.1
Se han señalado semejanzas en lo que respecta a la acumulación de proteína amiloide en un grupo de enfermedades entre las cuales se encuentran la EA y las enfermedades priónicas agrupadas bajo la clasificación de enfermedades conformacionales, aunque las alteraciones proteicas estructurales en el orden del plegamiento son diferentes como disímiles sus expresiones clínico-patológicas.2
Hasta el presente, sólo algunas enfermedades por priones tienen probada su infectividad siguiendo patrones clínicos particulares; desde que Prusiner identificó y definió el término prión,3 numerosos investigadores han señalado sólo un número limitado de esta “élite” entre las cuales se encuentran la enfermedad de Creutzfeldt-Jakob (ECJ), algunas que afectan al ser humano y otras formas de encefalopatías del ámbito veterinario, algunas de las cuales son transmisibles a la especie humana.4,5 Las enfermedades conformacionales2,6 pudieran ser un eslabón entre las alteraciones estructurales y la adquisición de cierto grado de infectividad en las enfermedades priónicas y la EA.
DESARROLLO
Los cambios en la estructura proteica incluyen un proceso de nucleación, en el cual existen oligomeros activos que funcionan como una "semilla" para convertir una conformación normal en la isoforma anormal, estas proteínas con trastornos en el plegamiento son producidas con frecuencia en el citoplasma o el retículo endoplásmico, este proceso está ocurriendo de forma continua en la célula, donde sucede una degradación proteosomal, existen proteínas que actúan previniendo la acumulación de aquellas mal plegadas y el desarrollo de enfermedades por esta causa.
Al fallar estos mecanismos, ocurre acumulación de proteínas no plegadas prefibrilares que forman agregados intracelulares y resultan citotóxicos.7,8 Tal vez, la presencia en cuanto a calidad o cantidad de estos agregados prefibrilares intervienen de forma directa en la capacidad para tornar una proteína normal en infecciosa y potencialmente contagiosa, esto se ha comprobado en cierto grado en la amiloidosis sistémica,9 lo que parece indicar la relación entre los cambios conformacionales y el carácter infeccioso de algunas proteínas; trabajos recientes sugieren que las enfermedades por priones pueden ser consideradas como una forma transmisible de amiloidosis cerebral.2,9,10 Otros autores sin embargo, no lo consideran así porque no resulta claro si los agregados presentes en estas entidades están implicados en la neurodegeneración de forma directa o si sólo son una consecuencia de la misma.2
El término prión tendría sentido como adquisición del carácter infeccioso de una proteína celular y pudiera ser empleado como sinónimo de transformación o cambio proteico infeccioso.
Aunque la proteína amiloide es diferente en muchos casos en cuanto a aspecto, tamaño, secuenciación o estructura nativa, todas ellas forman agregados fibrilares que muestran un marcado parecido en cuanto a morfología, estructura interna y a la presencia de aquellas proteínas llamadas serpinas, las cuales son un elemento común entre virus, células eucariotas y procariotas.11 Cambios en el pH, temperatura o exposición a ciertos agentes químicos pueden producir rupturas y cambios en la proteína original para formar proteína amiloide y, en muchos casos, estructuras fibrilares.12
El depósito de proteínas anormales con una u otra conformación produce enfermedades con deterioro cognitivo, trastornos conductuales, convulsiones y tienen un curso fatal; en el orden anatomopatológico se ha observado la acumulación de proteínas anormales en cuanto a su conformación y función, formación de placas amiloides y ovillos neurofibrilares y cambios degenerativos al nivel del núcleo de Meynert.13-15
Se desconoce a ciencia cierta hasta qué punto la EA y las llamadas enfermedades priónicas representan un peligro en el orden epidemiológico-infeccioso, es posible que constituyan en el futuro epidemias devastadoras por sus mecanismos de transmisión y su carácter solapado.
En el caso de las enfermedades priónicas como la ECJ, se invocan las mutaciones peptídicas de tal forma que se produce una estructura beta plana que ha sido demostrada como infecciosa, la llamada proteína priónica.1,15,16
Cualesquiera que sean los mecanismos mediante los cuales estas proteínas producidas se comportan como infecciosas, la no demostración del carácter definitivamente contagioso de la EA hasta este momento, pudiera deberse al largo período de latencia que ocurre desde la adquisición de la proteína infecciosa hasta el desarrollo de los primeros síntomas o signos de la enfermedad en el orden clínico, los cuales, por lo general, demoran años en manifestarse, si en un inicio las enfermedades producidas por priones fueron llamadas enfermedades por virus lentos17 fue precisamente por lo tardío de la expresión clínica con respecto al momento de la adquisición del contagio, ahora, con respecto a la EA, ¿acaso no pudiera tratarse de otra enfermedad producida por "virus más lentos"? usando los antiguos y ya caducos términos para una mejor comprensión del problema. Por otra parte, la coexistencia de demencia de causa vascular con la EA pudiera enmascarar los síntomas iniciales de esta última; además muchos ancianos mueren sin que se les realice un estudio mas allá del rutinario para establecer tal diferencia.
La incidencia de la EA en el mundo entero pudiera ser mucho más grande que lo que ha sido publicado, pues hay numerosos países que no contemplan este tema entre los objetivos priorizados de su sistema de salud.18-20
El surgimiento del concepto de proteína amiloide infecciosa dentro del contexto de las enfermedades conformacionales con expresiones clínicas diversas pudiera ser de importancia, pues ello pudiera incluir lo que de forma más estrecha se llama proteína priónica en las enfermedades por priones conocidas o proteina tau en la EA.
Quizá los cambios proteicos infecciosos sean diferentes tipos de respuestas de las proteínas del sistema nervioso a una gama limitada de agentes causales, determinando su expresión en el orden clínico-patológico y según las particularidades del código genético que les da origen, manifestándose unas veces como cualquiera de las enfermedades priónicas conocidas y otras veces de manera diferente como la EA, aunque esta hipótesis parece ser menos probable.
En un ordenamiento evolutivo decreciente, de los hongos a las bacterias, de estas a los virus y de estos últimos a los cambios o transformaciones proteicas infecciosas que pudieran constituir el elemento más primitivo de esta secuencia, parecen existir leyes que rigen el comportamiento y capacidad de cada escalón para producir enfermedades, pues mientras más primitiva es su estructura, mayor es el daño biológico que causan, más confusos resultan sus mecanismos productores de daño, más difíciles son de reconocer epidemiológicamente debido al poder de mutabilidad de unos o el mimetismo de otros, los cuales llegan al extremo de parecerse a proteínas celulares normales y evadir el sistema inmune quien no los reconoce en muchos casos como extraños aún habiendo variado su conformación.
¿Pero acaso proteínas aparentemente inocuas, tienen la capacidad de transformarse en infecciosas bajo determinadas condiciones o son un grupo de ellas las que lo hacen? La respuesta a esta interrogante se desconoce aún, pero pudiera ser un buen punto de partida para investigaciones posteriores.
En conclusión, la adquisición del carácter infeccioso de una proteína celular pudiera deberse a cambios en su conformación en relación con aspectos ambientales y genéticos, tanto para la EA como en las enfermedades priónicas, la carencia de pruebas del carácter infecto-contagioso de la EA tal vez se deba a su largo período de latencia y a lo confuso de sus diferencias con otras formas de demencia, es posible que las transformaciones proteicas infecciosas sean un eslabón evolutivo dinámico, pero inferior en relación con los virus, hongos y bacterias, de ahí la variabilidad en su expresión clínica.
REFERENCIAS BIBLIOGRÁFICAS
1. Velayos JL, Irujo A, Cuadrado-Tejedor M, Paternain B, Moleres FJ, Ferrer V. The cellular prion protein and its role in Alzheimer's disease. Prion. 2009;3:110-7.
2. Lupi O, Peryassu M. An emerging concept of prion infections as a form of transmissible cerebral amyloidosis. Prion. 2007;1:223-7.
3. Prusiner SB, Hsiao KK. Human prion diseases. Ann Neurol. 1994;35:385-95.
4. Zivkovic S, Boada M, López O. Revisión de la enfermedad del Creutzfeldt-Jakob y otras enfermedades priónicas. Rev Neurol. 2000;31:1171-9.
5. Biacabe AG, Jacobs JG, Bencsik A, Langeveld J, Baron T. H-type bovine spongiform encephalopathy. Complex molecular features and similarities with human prion diseases. Prion. 2007;1:61-8.
6. Mena M, Rodríguez-Navarro J, García de Yébenes J. The multiple mechanisms of amyloid deposition. The role of parkin. Prion. 2009;3:5-11.
7. Stefani M, Dobson CM. Protein aggregation and aggregate toxicity: new insights into protein folding, misfolding diseases and biological evolution. J Mol Med. 2003;81:678-99.
8. Gregersen N, Bross P, Andresen BS, Pedersen CB, Corydon TJ, Bolund L. The role of chaperone-assisted folding and quality control in inborn errors of metabolism: Protein folding disorders. J Inherit Metab Dis. 2001;24:189-212.
9. Lundmark K, Westermark GT, Nystrom S, Murphy CL, Solomon A, Westermark P. Transmissibility of systemic amyloidosis by a prion-like mechanism. Proc Natl Acad Sci USA. 2002;99:6979-84.
10. Maddelein ML. Infectious fold and amyloid propagation in podospora anserina. Prion. 2007;1:44-7.
11. Belorgey D, Hägglöf P, Karlsson-Li S, Lomas D. Protein Misfolding and the Serpinopathies. Prion. 2007;1:15-20.
12. Dickson DW, Crystal HA, Bevona C, Honer W, Vincent I, Davies P. Correlations of synaptic and pathological markers with cognition of the elderly. Neurobiol Aging. 1995;16:285-98.
13. Jahn TR, Radford SE. The Yin and Yang of protein folding. FEBS J. 2005;272:5962-70.
14. Guimerà A, Gironès X, Cruz-Sánchez F. Actualización sobre la patología de la enfermedad de Alzheimer. Rev Esp Patol. 2002;35:21-48.
15. Ariza A. El patólogo ante las encefalopatías espongiformes transmisibles. Rev Esp Patol. 2002;35:49-62.
16. Hamilton JA, Whitty G, White AR, Jobling MF, Thompson A, Barrow CJ. Alzheimer's disease amyloid beta and prion protein amyloidogenic peptides omote macrophage survival, DNA synthesis and enhanced proliferative response to CSF-1 (M-CSF). Brain Res. 2002;940:49-54.
17. Fernández MI, Arribas MA, Fernández Buey N. Demencias por virus lentos: enfermedades tipo prión (encefalopatías espongiformes transmisibles). Medicine. 1998;7:4470-3.
18. Alzheimer's Disease Education and Referral Center. Progress report on Alzheimer's disease: taking the next steps. Silver Spring, MD: 2001. (NIH Publication No. 00-4859).
19. Petersen RC. Practice parameter: early detection of dementia: mild cognitive impairment (an evidence-based review), Report of the Quality Standards Subcommittee of the American Academy of Neurology. Neurology. 2001;56:1133-42.
20. National Center for Health Statistics. Deaths and death rates for the 10 leading causes of death in specified age groups, by race and sex: United States, 1998. National Vital Statistics Reports. 2000;48(11):27-9.
TITULO: Prion, ¿partícula proteica estable o transformación dinámica? Un nuevo enfoque sobre las enfermedades priónicas y la enfermedad de Alzheimer
AUTOR: Edmundo Rivero Arias. Especialista de I Grado en Neurología. Investigador Auxiliar. Profesor Auxiliar. Hospital Clinicoquirúrgico "Hermanos Ameijeiras". La Habana, Cuba.
CITA BIBLIOGRÁFICA: Prion, ¿partícula proteica estable o transformación dinámica? Un nuevo enfoque sobre las enfermedades priónicas y la enfermedad de Alzheimer. Revista Cubana de Medicina. Versión impresa ISSN 0034-7523. Rev cubana med v.49 n.3 Ciudad de la Habana jul.-sep. 2010. [En Línea]. http://scielo.sld.cu/scielo.php?pid=S0034-75232010000300005&script=sci_arttext. 07 de Marzo de 2011.
PREGUNTA DE INTERÉS
¿Pero acaso proteínas aparentemente inocuas, tienen la capacidad de transformarse en infecciosas bajo determinadas condiciones o son un grupo de ellas las que lo hacen?
lunes, 28 de febrero de 2011
Información de Priones.
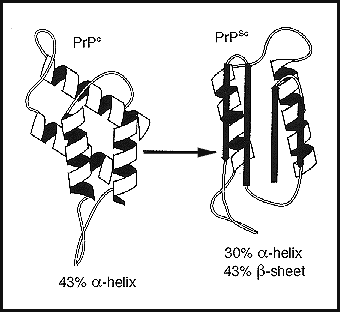
Características de los priones
Una vez obtenido el prion responsable de la patología, comenzó a dilucidarse su estructura. Se trata de una glicoproteína de 27-30 kD, que tiene la misma estructura primaria que una proteína similar presente en el cerebro de la oveja. La modificación estructural, no genética, se debe a un proceso de postraducción. Esto se presenta en funciones muy especiales. El gen codificando del prion Prpc (gen PRNP) se localiza en el brazo corto del cromosoma 20, no tiene intrones y es un gen autosómico dominante. Se expresa en el tejido neuronal, cardíaco, muscular, pancreático y hepático. El plegamiento erróneo de la Prpc a PrpSc confiere a la PrpSc dos propiedades que la diferencian de la Prpc: la resistencia parcial a la digestión por proteases y su insolubilidad. Estas dos propiedades hacen que la PrpSc sea estable y la capacitan para poder formar agregados proteicos responsables de la acumulación de PrpSc en forma de placas amiloides en el tejido nervioso. [6] Las principales diferencias entre la forma normal del prion, Prpc , y la forma patógena, PrpSc son las siguientes:| Prpc | PrpSc |
|---|---|
| Estructura hélice α (4 regiones de proteína globular) | Estructura lámina β (proteína plana) |
| Susceptible a proteasas | Resistente a proteasas |
| Proteína monomérica | Agregados proteicos |
| Monómeros estables | Monómeros poco estables (agregados amiloides) |
| Resistencia normal | Resistencia extrema a la radiación y disolventes fuertes |
| Soluble en detergentes | Insoluble en detergentes |
Es importante destacar que tanto las Prpc como las PrpSc tienen la misma secuencia de aminoácidos.
Tomado de: http://es.wikipedia.org/wiki/Prion
Este video fue tomado de: youtube
Preguntas frecuentes:
- ¿Qué son los priones?
- ¿Cuál es su estructura molecular?
- ¿ Cuál es su función en los seres vivos?
- ¿Qué enfermedades son causadas por priones?
Suscribirse a:
Entradas (Atom)